Challenge Overview
Problem Statement | |||||||||||||
Prizes$20,000 in prizes divided among the top 5 finishers:
BackgroundThe Connectivity Map (CMap) is a Broad Institute project that enables the discovery of functional connections between drugs, genes and diseases through the generation and analysis of gene expression signatures (see the papers in Science and Nature Reviews Cancer). Recent technological advances and laboratory automation have enabled data generation scale-up resulting in a massive catalog of drug and gene signatures. When coupled with powerful pattern recognition algorithms, these data represent a rich reference library for the research community. CMap utilizes a novel, high-throughput gene expression profiling technology to generate gene expression profiles at scale. The crux of this approach is that instead of measuring all ~20,000 genes in the human genome, CMap uses an assay called L1000 to measure a select subset of approximately 1,000 genes and uses these ���landmark��� gene measurements to computationally infer a large portion of the remainder, taking advantage of the fact that some genes��� expression levels are correlated via functional redundancy and/or shared regulatory mechanisms. The current algorithm is effective but imperfect, and improving the imputation methods will have an immediate impact on the quality of data and the biologically meaningful connections that can be discovered. With this in mind, we have designed our first contest to stimulate the exploration of new and improved inference methods. The existing gene inference CMap approach uses an ordinary least squares regression model. That is, each non-landmark or ���inferred��� gene is estimated as a linear combination of all landmarks. Linear models are popular across a variety of domains and can be built easily using even small training datasets. The CMap inference model was trained using a compendium of gene expression data generated across a diversity of human tissue types and cell lines. This approach makes the assumption that the relationships between the landmark and inferred genes are linear and that they are generally conserved across tissue types and cell lines. In theory, utilizing alternative computational approaches that model nontrivial relationships and leverage larger compendia of data, could further improve the predictions. This contest seeks to test that hypothesis. Problem DescriptionYou are given a large training dataset (100,000 samples) of 12,320 gene expression levels that include 970 landmark and 11,350 non-landmark genes. (The existing CMap model was trained on a very similar dataset.) You are also provided with a smaller offline-testing dataset of 1,000 samples where the landmark and non-landmark genes are in separate files (for compatibility with the scoring scripts). The challenge is to build a model that uses the 970 landmark genes to predict the expression levels of the inferred genes for a distinct set of 1,650 separately measured samples. Additional details about the scoring are provided below. Data DescriptionData FormatData is provided as CSV files where the rows correspond to genes and the columns correspond to samples. The matrix values are floating point numbers in the range 0.00 to 15.00. The first few rows and columns of one such file is shown in Figure 1 below. 6.85,4.78,7.83,8.37,4.57,10.91,14.55,8.24,6.23,8.43,10.14,8.21,8.15,7.29,13.07,9.86,9.86,5.44 7.98,11.37,8.1,7.19,5.52,7.99,10.41,8.09,6.8,9.24,8.81,8.67,8.29,8.49,10.52,9.71,9,6.46,8.37,6.73 5.68,4.27,7.49,8.44,5.18,7.17,9.24,7.81,4.59,7.76,9.08,5.94,7.43,7.86,8.04,9.22,10.08,7.14,6.91 9.52,7.82,9.2,7.03,5.42,8.11,10.05,7.98,6.95,7.96,11.91,7.62,8.62,8.27,10.6,8.4,8.42,6.64,9.26,7.11 6.37,4.42,10.31,8,6.49,6.19,11.17,7.11,6.61,6.8,10.71,4.62,4.43,9.38,7.78,11.34,7.7,5.83,7.56,7.22 9.56,6.27,8.94,6.27,7.47,7.77,8.38,12.45,7.03,6.31,7.27,4.22,10.74,10.91,8.65,4.47,6.51,7.87,7.24 6.68,10.56,8.7,7.95,5.97,6.92,9.67,9.36,7.95,8.1,8.64,10.11,4.42,10.99,9.62,6.21,7.18,7.74,10,7.28 10.54,5.66,8.61,6.38,6.13,9.61,7.28,8.76,7.62,6.67,12.58,6.81,6.18,8.91,9.37,7.01,7.02,8.68,12.93 7.08,4.74,8.64,7.72,5.59,7.73,9.99,6.93,6.36,7.38,8.87,7.5,7.72,9.12,11.32,7.84,8.34,6.26,9.04,6.25 7.79,6.84,8.51,6.76,6.09,7.72,9.64,7.92,5.96,6.81,10.68,6.08,9.11,7.97,10,8.94,7.33,6.47,8.03,7.06 Figure 1. Example CSV matrix format. These matrices are headerless CSV files with genes along the rows and samples along the columns. Training Datatraining.csv: 100,000 samples of 12,320 genes. The first 970 rows are the landmark genes and the last 11,350 are the non-landmark genes. Offline Testing Data
Test DatatestData.csv:
1,650 samples with the landmark genes measured by L1000 MethodsThe objective is to predict the non-landmark genes��� expression values for the 1,650 samples in testData.csv. Your output should be formatted as a CSV file and should contain exactly 11,350 lines corresponding to each non-landmark gene. Each line should contain 1,650 numerical values representing your predicted expression levels for each sample. The rows and columns of the data should be in the same order as they appear in the provided test data. To submit your entry, your code will only need to implement a single method, getURL(), which takes no parameters, and returns the URL at which your predictions CSV file can be downloaded. Example TestingAll example testing for this contest should be done offline, using the provided data. Note, however, that you may do a ���Test Examples��� submission using your predictions file for the provided test data. This will not provide any provisional scoring, but will confirm for you that the predictions file works correctly (URL is accessible, correct number of rows and columns, and numerical values parse correctly). This is not required, but can be used as a basic sanity check before making a full submission. Scoring OverviewThe scoring function is designed to reward improved performance relative to the existing CMap inference model. For each predicted gene, the scoring function takes into account two metrics of imputation performance:
Those two metrics are averaged to arrive at a single score for each inferred gene. The gene-level scores for your algorithm will be compared to the equivalent scores that have been pre-computed using the existing CMap inference model.
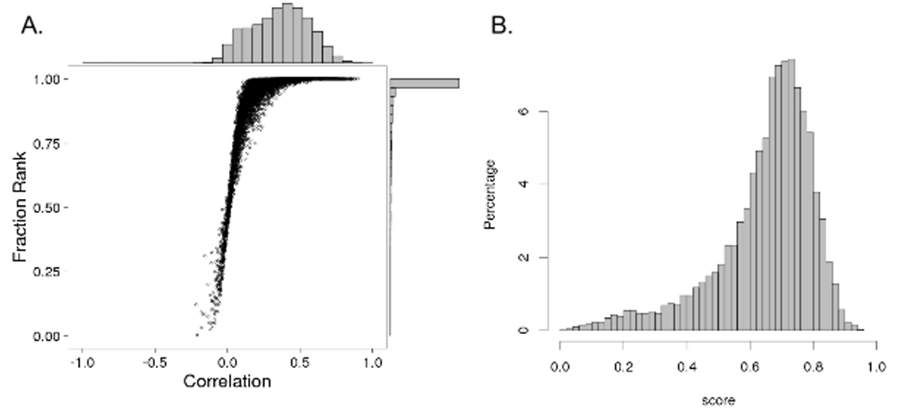
Submissions will be scored based on the algorithm���s ability to generate the expected expression patterns for each inferred gene across the M samples in question. You are provided with an R script and a java executable that both contain an implementation of this scoring algorithm that you can use for offline evaluation of your model. Algorithms doing no better than the current model will receive scores of approximately 1,000,000. Scores above 1,000,000 indicate algorithms doing better than the existing model and scores below 1,000,000 indicate algorithms doing worse. Running the Scoring ScriptsThe R scoring script can be invoked as follows: Rscript cmap_scoring_function.R --inf_ds [/path/to/inference/result.csv] \
--truth_ds [/path/to/truth/matrix.csv] \
--reference_scores [/path/to/reference/scores.csv] \
--out [/desired/output/path]
You can also run `Rscript cmap_scoring_function.R -h` to view the script���s help message. Running the Java executable: java ConnectivityMap [prediction_file] [truth_file] [ref_score_file] Note that the Java scorer will simply output the computed score total, without rescaling against the maximum possible score. However, it is possible to calculate ScoreMax for any set by testing the ground truth against itself. (See details below) Scoring Algorithm DetailsThe scoring will be computed as a Spearman correlation of the gene expression values, ranked across the samples, with the ground truth. Let S be a matrix N * M of gene expressions, output by your algorithm, where N = totalCases is 0he number of samples in the testcase and M = 11350 is the number of inferred genes. Let G0to be the corresponding matrix N * M of gene expression measures (i.e. ground truth). Let S(j) be a list of expressions across the sample of the gene for j in (1..M). Let G(j) be the corresponding list from ground truth. Then, a raw single-gene score is computed as as a mean of the correlation across the samples and it���s scaled rank across the genes: ScoreRaw(j) = 1/2 * ( CorrelationScore(j,j) + (1/M) * Ranked[CorrelationScore(j,All)](j) ) Where the correlation matrix is CorrelationScore(j,i) = Correlation [ Scaled [ Ranked [ S(j) ] ], Scaled [ Ranked [ G(i) ] ] and CorrelationScore(j,All) is the vector of correlations between inferred gene j and all ground truth genes. and the functions Correlation, Scaled and Ranked are: Correlation [ X, Y ] = Total [ X * Y ] / (N - 1) ; Correlation [ X, Y ] -> 0, if Correlation [ X, Y ] <0 Scaled [ X ] = (X - Mean [ X ]) / StandardDeviation [ X ] Scaled [ X ] -> 0 * X, if StandardDeviation [ X ] = 0 Ranked returns a position the element of the list would have if the list was sorted in increasing order. If several elements on the list have the same value, the averaged position for those elements is returned. To illustrate: Ranked [ (2, 1, 0, 4.1, 9.5, 5, 5) ] = (3, 2, 1, 4, 7, 5.5, 5.5) For the final score, the raw single-gene scores are normalized using the benchmark score (BenchmarkRaw(j) for j in (1..M)) for each of the genes, produced by a linear-regression model, averaged across the genes, and rescaled with the use of maximal score achievable on the test (i.e. the score one would obtain with perfect predictions): ScoreFinal = Max (0, ScoreTotal * (ScoreMax - 1,000,000) / (ScoreMax - ScoreTotal)) Where ScoreTotal = 1,000,000 * Mean [ (2 - BenchmarkRaw) / (2 - ScoreRaw) ] And ScoreMax is computed as ScoreTotal when a ground truth is substituted as an answer. ScoreMax has been computed separately for provisional and system tests and is being kept as a built-in constant for each of those. Requirements to Win a Prize
| |||||||||||||
Definition | |||||||||||||
| |||||||||||||
Examples | |||||||||||||
| 0) | |||||||||||||
| |||||||||||||
This problem statement is the exclusive and proprietary property of TopCoder, Inc. Any unauthorized use or reproduction of this information without the prior written consent of TopCoder, Inc. is strictly prohibited. (c)2020, TopCoder, Inc. All rights reserved.