Challenge Overview
Problem Statement | ||||||||||||||||||||||||
IntroductionThe Connectivity Map (CMap) is a Broad Institute project that enables the discovery of functional connections between drugs, genes and diseases through the generation and analysis of gene expression signatures, where each signature represents the transcriptional responses of human cells to chemical or genetic perturbations. To identify such connections, a CMap user poses a biological question in the form of a ���query��� comprising a list of genes of interest. The query algorithm then searches the CMap database to identify the signatures that are most similar to the user's input. The biomedical community has used CMap to discover relationships between drugs, diseases, and genes and to generate new scientific hypotheses that point the way to therapeutic solutions. An example use case is as follows: A researcher discovers a set of genes that are collectively dysregulated in a certain disease. She then queries CMap with this gene set and discovers a drug that inversely matches her query and therefore hypothesizes that the drug may be of therapeutic relevance for the disease. The CMap assay quantifies the responses of 10,174 genes to an experimental perturbation, such as a drug. Due to recent technological improvements, it has become possible to massively scale-up data generation and as a result the CMap signature database has grown to 476,251 signatures and this number is expected to increase rapidly. Hence improving the speed of the query algorithm is of great practical importance to those seeking to use CMap for therapeutic discovery. The goal of this contest is to reduce the time the query algorithm takes to execute. Support for the Connectivity Map project comes from the NIH Library of Network-Based Cellular Signatures (LINCS) and NIH Big Data to Knowledge (BD2K) Programs. The aim of the LINCS program is to understand biological systems by profiling gene expression and other cellular processes when exposed to perturbagens. The goal of the BD2K program is to support research aimed at accelerating the use of Big Data applications and tools in biology. The Connectivity Map project is grateful for their support. If you would like to access datasets for past CMap contests, or would like to learn more about CMap, please visit their website. Prize Distribution
Note: As part of the conditions of winning a prize, contestants will be required to submit a written report documenting the approach of how their code works. Competitors will optionally be allowed to provide their final submission for system testing in the form of an AWS VM (or AMI) with a standalone version of their solution running on it. While this is not required, it is encouraged, particularly for those competitors wishing to use multi-threaded solutions and/or take advantage of low-level configuration of how their code runs. (e.g. anything outside of the capability of testing when on the typical marathon platform) Problem DescriptionThe query algorithm starts with an a priori defined pair of gene sets. Typically, these are user-defined sets of genes that are up- and down-regulated under a biological condition of interest. The goal of the query process is to rank order all signatures within the CMap signature matrix by similarity to the pattern of up- and down-regulated genes specified in the query gene set. Similarity between query gene set q (which can range in size from 10 to a few hundred genes) and a signature s (which is a vector of 10,174 values, one per gene, where each value is a measure of differential expression and therefore indicates the magnitude of the given gene���s response to the perturbation) in the CMap signature matrix (M genes x S signatures) and is computed using the weighted Kolmogorov-Smirnov (wtks) statistic, as described in Subramanian, et al., which reflects the degree to which genes in q are overrepresented at the top or bottom of the signature s. The wtks is computed separately for the up and down gene sets in the query q and then integrated into a combined wtks (wtkscomb). For a query q, this procedure is iteratively applied to each of the 476,251 signatures S (columns) of the CMap signature matrix, resulting in a rank ordered wtkscomb list. This process is easily extended to multiple query gene set pairs Q, in which case the query output is a matrix of 476,251 signatures S as rows by Q columns. Description of the algorithmFor a given query gene set q containing N genes, and a signature of differential expression scores s for M genes, the single-sided wtks (wtksss) value is computed as follows:
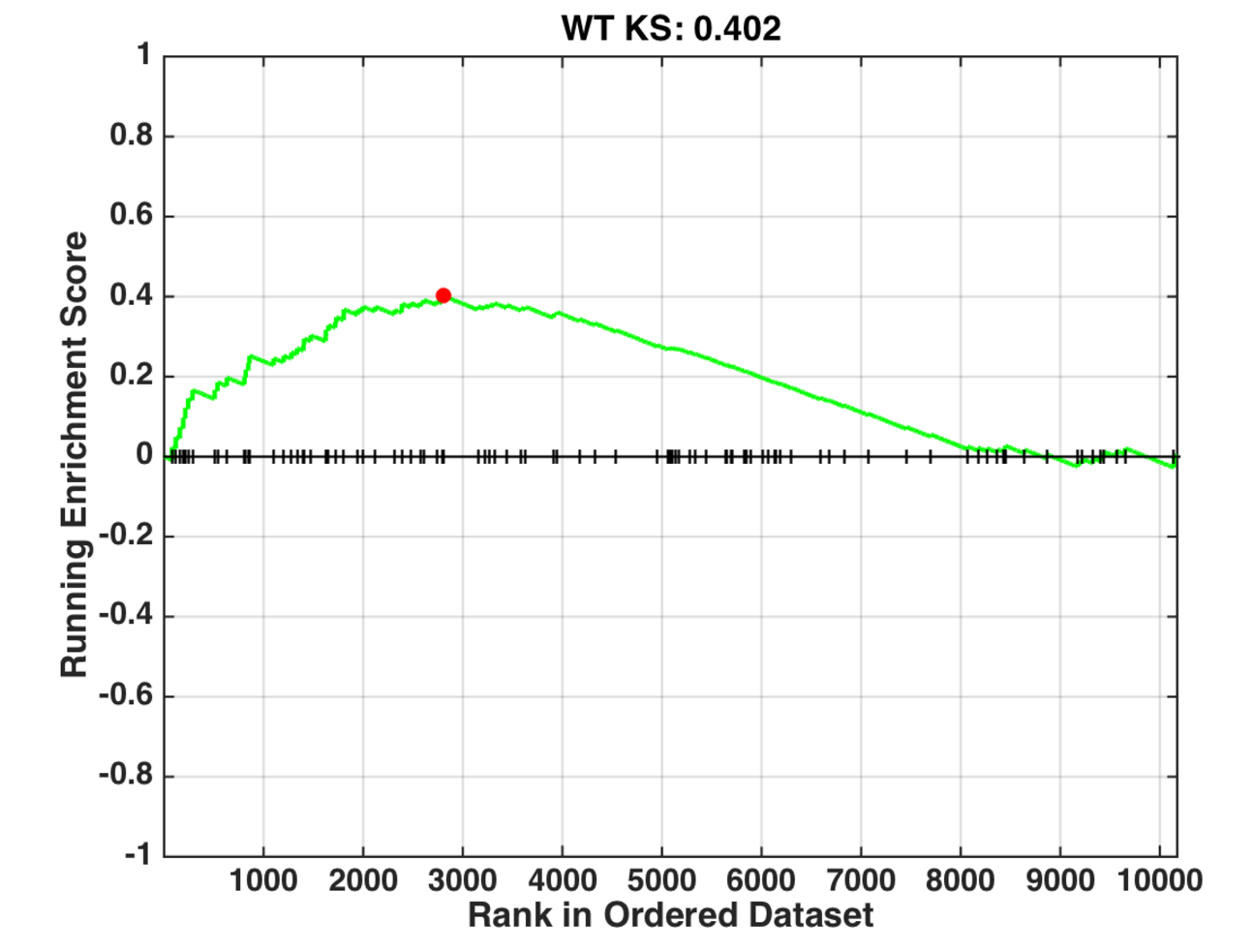
The ���mountain��� plot below depicts the RS for an example query gene set q and an example signature s. The vertical ticks on the horizontal line at y = 0 denote positions in s that correspond to genes in q. The red dot indicates wtksss, the maximum deviation from zero.
CMap queries are comprised of a paired up and down gene set (qup and qdown). We leverage this to compute a combined wtks (wtkscomb) that integrates the single-sided wtks for both the up and down gene sets. wtkscomb is computed as follows:
+---------------------------+-------------------------+ | wtksup and wtksdown signs | wtkscomb | +---------------------------+-------------------------+ | same | 0 | +---------------------------+-------------------------+ | different | (wtksup - wtksdown) / 2 | +---------------------------+-------------------------+ Example CodeAvailable for download here. Contestants are provided with example Matlab code that implements the wtkscomb algorithm as described above. Along with this code, we also provide a toy dataset, on which the code can be run, to illustrate the mechanics of the algorithm and the desired output format. The specific files provided are:
Note that this code is also compatible with Octave, an open-source statistical environment similar to Matlab. Current optimization strategiesThe CMap group has adopted a few optimizations to speed up queries. These are enumerated here so as to describe past work and provided to contestants as potential avenues for further improvements:
Contestants are allowed to improve upon the above approaches���however simply implementing the above is unlikely to yield sufficient improvement over baseline performance as they are already in the current implementation. Contestants might also explore other approaches including more efficient code (restricted to languages Java, C++), more performant file i/o (note that the test environment loads data off local SSD-based storage), using CPU-level multiprocessing etc. All VM-based tests will be run on an Amazon EC2 instance of the following specification and contestants are allowed to use all available resources on the instance: +---------------+--------------+-------+------------------+ | Instance Type | Memory (GiB) | # CPU | SSD Storage (GB) | +---------------+--------------+-------+------------------+ | c3.4xlarge | 30 GB | 16 | 2 x 160 | +---------------+--------------+-------+------------------+ * The instances used for testing on the platform during the match may not match the above, but all submissions will be scored on identical instances. The instance type above will be used for all post-contest validation. File typesGene sets are provided as CSV files. These are comma-delimited text files where each row contains a single gene set. The first and second columns contain gene set IDs and descriptions, respectively, and the remaining columns contain the genes in the gene set. For this contest, the description column is empty for all gene sets. Each row can contain values for an arbitrary number of columns. For a given gene set collection, there are two CSV files, one each for the up and down gene sets. It is assumed that the sets are in the same order in both the up and down files. * When your getWTKScomb() method is called during the contest, there will be no heading rows or columns. Each line of the input will be a comma delimited list of genes for each query. Matrices are provided as CSV files with row and column identifiers. Contestants' code should output CSV files of 476,251 rows by N columns, where N is the number of query gene set pairs for a given collection. * During the contest your code should return a double[] with 476251 * N values, where the first N elements represent the wtkscomb for each query against the first signature, and so on. Useful linksl1ktools: Github repository containing code to parse and interact with various CMap file types, including GCTX. Method ImplementationYou will need to write several(*) methods: int init(int[] geneList) This method will provide your code with the list of 10174 gene IDs used during the contest, in the order in which they appear in all of the data. This method is free to pre-load and/or pre-index data, including the saving of scratch files. However, the genes which will be part of the queries are not yet provided. This method is not timed for scoring purposes (although there is a limit to the total allowable runtime). double[] getWTKScomb(string[] up_collection, string[] down_collection) Given a collection of pairs of up and down gene sets, compute the wtkscomb for all signatures (columns) in the CMap signature matrix. The execution speed of this method is timed, and the results judged according to their accuracy, as explained below. Available Library Methods (Class CMAPLib)Several methods are provided in the CMAPLib class for your code, which you can call to save and load scratch files during the test process. They each function in the obvious way. int saveIntFile(String filename, int[] data) int saveDoubleFile(String filename, double[] data) These methods will save out scratch files for later use during the test run. The return values are 0 in the case of success of -1 in case of error. int[] loadFromIntFile(String filename, long start, int length) double[] loadFromDoubleFile(String filename, long start, int length)
Data Files ProvidedFour data files are provided by default, and are available on the test machine to be loaded from your code:
TYPES OF TESTINGOffline testing (does not contribute to scores, for development purpose only). Contestants are provided with query gene set pairs and the CMap signature score and rank matrices. These files are intended to be used for building and testing wtkscomb implementations. These files include:
Copies of the binary files used on the tester are available here (note that the values are identical to what is in the CSV files, just converted to a binary format to facilitate loading): Provisional testing (populates the leaderboard). During the match, contestants have access to the full CMap Signature Matrix (as in binary format above, which contains the same data as CSV format sig_score_n476251x10174.csv and sig_rank_n476251x10174.csv) along with the Provisional Query Sets (500 pairs of up and down gene sets, each set has a variable number of genes). System testing (used for holdout analysis and final selection of winners). At the conclusion of the match period a contestant's submission will be evaluated against a collection of 1,000 holdout Query sets as a measure to ensure the Provisional result was not overfit. Importantly, the CMap signature matrix used in holdout testing will be identical to that used in Provisional, but the Query sets will be entirely different. As mentioned earlier in the problem statement, competitors will have the option to have their code tested "bare-metal" (well, bare-VM) on an image they setup to run standalone. Though this is not required, it is highly recommended for those wishing to implemented multi-threaded solutions or take advantage of low-level tweaking. Notes on standalone submissions: Your code should be setup so it can be run from a single
command: "./run.sh <upfile> <downfile> <outputfile>" ScoringAccuracy: As this contest is focussed on improving speed, we seek solutions that are exactly the same numerically as the reference algorithm. We compute the absolute differences between a matrix of test and ground truth wtkscomb values according to the formula: D = abs(WTKScont - WTKSref) Where WTKScont is a matrix of contestant-generated wtkscomb values, WTKSref is the equivalent reference matrix, and D is a matrix of the absolute element-wise differences between WTKScont and WTKSref. All elements of D must be less than 0.001 to be considered sufficiently accurate and eligible for timing-based scoring. However, we recognize that some measures to improve speed, such as choice of data types and optimizing data storage by encoding score and rank in the same matrix, may result in small losses in precision. This in turn may cause stochastic changes in the sign of wtksup or wtksdn in cases where the gene ranks are randomly distributed and max and min of RS are very close in magnitude. As described above, if wtksup and wtksdn are both of the same sign, wtkscomb will be set to zero. This can lead to test values of wtkscomb differing from ground truth in a small percentage (<0.001%) of cases. In all such cases, the wtkscomb value in either the test submission or ground truth will be zero. To account for this, we allow for up to 1,000 such differences of greater than 0.001 between the test submission and ground truth where either the test or ground truth wtkscomb value is zero. Submissions that exceed this number will be disqualified. Timing: All algorithms that pass the accuracy constraints will be scored based on their time to execute on the entire database of gene sets provided. The goal is for the algorithm to execute as fast as possible, so faster executions will receive higher scores. The algorithm score will be based on the scoring function: Score = 1000000 * b / (b + 4 * t) Where t is the runtime of the submission and b is the baseline runtime of the existing algorithm. For the provisional test, b = 60 mins, and for the system test, b = 120 mins, respectively. Note that algorithm scores exceeding 200,000 indicate an improvement in speed over the existing algorithm, whereas scores less than 200,000 indicate no improvement. Notes on time calculation: The sandbox testing framework on the platform has a fair amount of overhead from serialization of passing data back and forth between the test harness and the competitor submission. The "runtime" reported when making example submission tests only includes the literal time spent in the competitor's submitted code. As disk I/O is part of this problem, the test harness internally keeps track of time spent processing file load/save functions once the query has been called (as above, time spent during the init() function is not tracked). These two values -- the submission runtime and the I/O time -- are summed together, and that sum is what is used as "runtime" for purposes of score calculation. Note specifically that time spent due to the data passing between the tester and the submission, is *not* counted in this time. | ||||||||||||||||||||||||
Definition | ||||||||||||||||||||||||
| ||||||||||||||||||||||||
Examples | ||||||||||||||||||||||||
| 0) | ||||||||||||||||||||||||
| ||||||||||||||||||||||||
This problem statement is the exclusive and proprietary property of TopCoder, Inc. Any unauthorized use or reproduction of this information without the prior written consent of TopCoder, Inc. is strictly prohibited. (c)2020, TopCoder, Inc. All rights reserved.